新闻网讯(通讯员笙柯)近日,细胞生物学国际期刊Autophagy(《自噬》)发表了武汉大学生命科学学院教授周荣家与程汉华课题组合作的最新研究成果。该研究揭示了线粒体NAD+调控线粒体自噬及维持细胞稳态的分子机制和功能。
论文题目为“Mitochondrial NAD+-mediated mitophagy alleviates type I interferon response to the cytosolic mitochondrial DNA”(《线粒体NAD⁺通过促进线粒体自噬减轻胞质mtDNA触发的I型干扰素反应》)。生命科学学院博士生兰天为第一作者,课题组博士生尚丹童、林岚、王昊宇、邹娟和胡萌欣参与了该项研究。周荣家和程汉华为共同通讯作者。
烟酰胺腺嘌呤二核苷酸(NAD⁺)作为细胞能量代谢的辅酶,通过参与氧化还原反应和调控代谢酶活性维持细胞稳态。线粒体NAD⁺分子的缺乏与人类各种疾病密切相关,包括自身免疫疾病、神经退行性疾病、心血管病、代谢疾病以及癌症等。因此,理解NAD⁺稳态机制、维持细胞中NAD⁺池的稳定对于人类健康至关重要。NAD⁺在细胞内呈现显著的区室化分布特征,其中线粒体NAD⁺的稳态调控尤为关键。线粒体自噬作为选择性清除受损线粒体的质量控制机制,在维持细胞稳态中发挥核心作用。然而,线粒体区室特异性NAD⁺是否调控线粒体自噬及其分子机制尚不明确。另一方面,线粒体自噬功能障碍可导致应激条件下线粒体DNA的异常泄漏,进而激活cGAS-STING信号通路并增强I型干扰素反应,但线粒体NAD⁺是否参与该过程的调控,仍有待阐明。
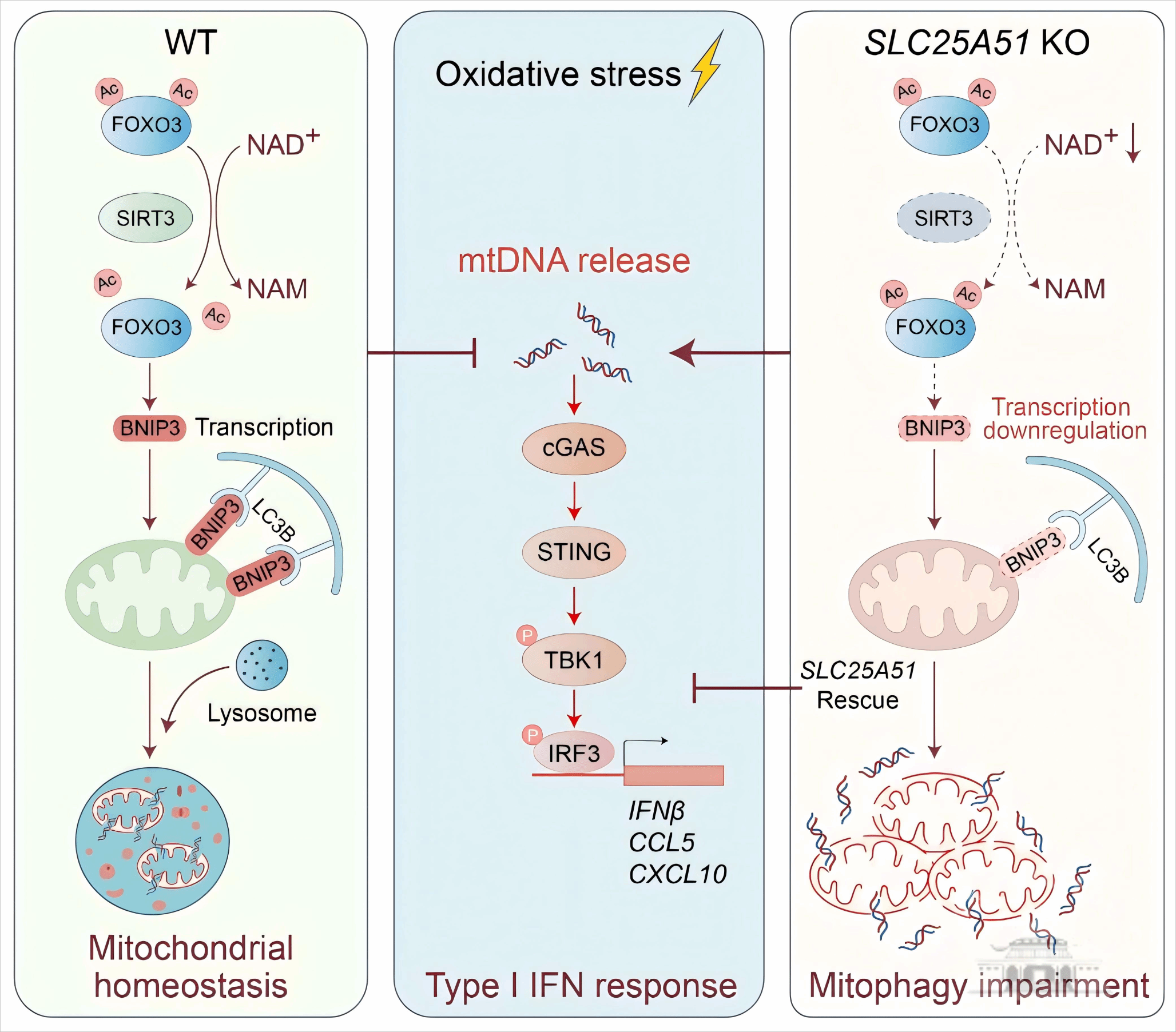
图1.线粒体NAD⁺转运蛋白SLC25A51通过调控线粒体自噬抑制线粒体DNA泄漏介导的I型干扰素反应
研究团队以线粒体NAD⁺转运蛋白SLC25A51为靶点,通过构建一系列基因敲除细胞模型,结合多重分子实验技术,系统解析了线粒体NAD⁺在调控线粒体自噬和炎症反应中的作用及机制。研究发现,SLC25A51缺失导致的线粒体NAD⁺水平下降会显著抑制线粒体自噬体的形成及其与溶酶体的融合,从而降低线粒体自噬通量,而回补SLC25A51可有效逆转这一缺陷。在机制层面,SLC25A51缺失削弱了SIRT3介导的FOXO3去乙酰化作用,导致FOXO3乙酰化与泛素化水平升高,进而加速其蛋白降解。FOXO3稳定性下降进一步减弱了其对下游基因BNIP3的转录激活能力,造成线粒体自噬受体BNIP3表达降低,最终引发线粒体自噬功能障碍。这一线粒体质量控制机制的紊乱,削弱了细胞对受损线粒体DNA的清除能力,在氧化应激情况下促使线粒体DNA向胞质泄漏。泄漏的线粒体DNA通过激活cGAS-STING信号通路,增强TBK1与IRF3的磷酸化,最终促进I型干扰素(IFNβ)以及炎症因子(CCL5和CXCL10)的表达与分泌。此外,临床数据分析显示,SLC25A51基因表达下调及其突变与人类自身免疫及炎症性疾病的发生显著相关。因此,这一发现揭示了NAD⁺代谢自噬炎症及自身免疫的分子关联机制(图1),为自身免疫及炎症性疾病的机制研究与靶向治疗提供了新的理论依据。
论文链接:https://www.tandfonline.com/doi/full/10.1080/15548627.2025.2589909
(供图:生命科学学院 编辑:相茹)